Findings:
Figure 1: Non-contrast CT shows a large rounded mass in the lateral ventricles with intermediate density and large foci of calcification within it. This is causing significant hydrocephalus with enlargement of the third ventricle.
Intraventricular oligodendroglioma
Oligodentrogliomas are well differentiated, slowly growing but diffusely infiltrating cortical and subcortical tumors. Although most of them involve the frontal and frontotemporal cortex, a small proportion of them are seen in the ventricular walls (1-10%), cerebellum and exceedingly rarely within the brainstem, spinal cord and leptomeninges. No age is exempt and the peak incidence is in the 4th or 5th decades. It usually have a long standing history of symptoms and the most common being seizures and headaches.
Intraventricular oligodentrogliomas can present with hydrocephalus. Pathologically, they are well defined, grayish-pink soft unencapsulated mass and calcification is extremely common. Focal cystic degeneration and hemorrhage are frequent findings. CSF seeding is uncommon. Histopathologically, these are moderately cellular tumors with occasional mitosis. Perinuclear halos or the "fried egg artifact" is a distinctive feature of oligodentroglioma. Majority of the "intraventricular oligodentrogliomas" described in the literature are central neurocytomas and immunohistochemistry helps to distinguish them.
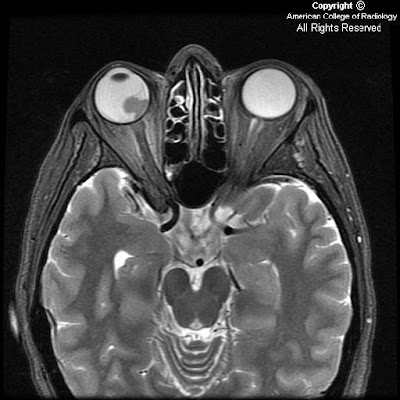
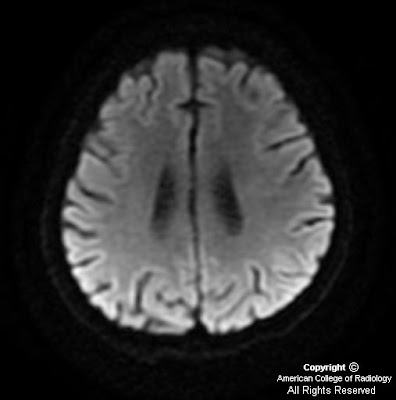
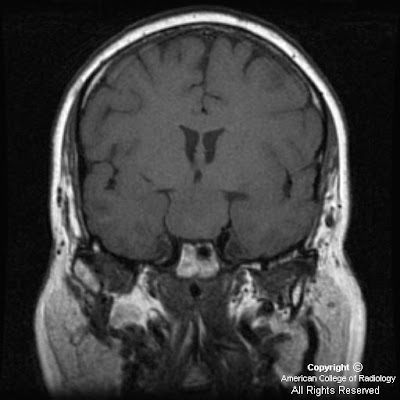
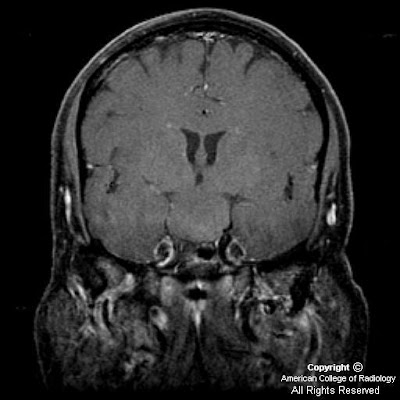
On CT, oligodentrogliomas usually have mixed density with nodular, clumped or linear tumoral calcification seen in majority (50-90%). Cystic degeneration is common. Intratumoral hemorrhage and edema is uncommon. The hemispheric lesions may expand, remodel or erode the calvarium. Enhancement is variable. On MR, oligodentrogliomas are typically heterogeneously hypointense or isointense to grey matter on T1 and heterogeneously hyperintense on T2 and FLAIR due to calcification, cystic change and hemorrhage. Nearly 50% show heterogeneous enhancement following contrast administration. Areas of calcification and hemorrhage demonstrate "blooming" on gradient echo MR sequences. No diffusion restriction on DWI.
Surgical resection is the primary treatment of choice. Radiation therapy and chemotherapy is reserved for recurrent tumors. Local recurrence is common and hence regular surveillance is recommended.